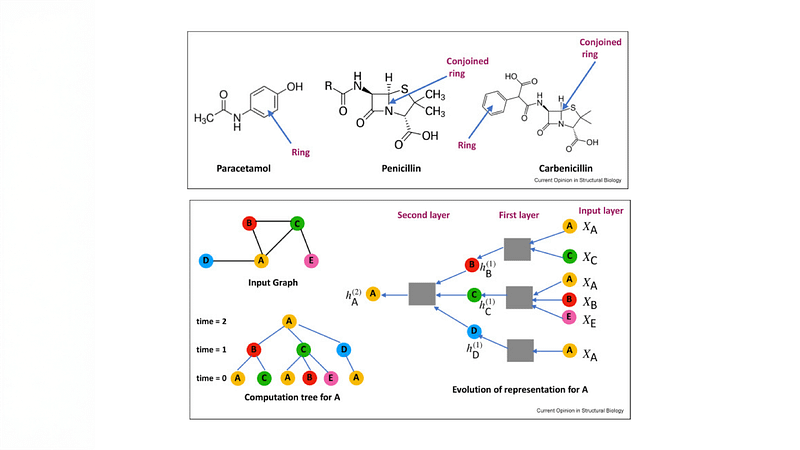
Graph neural networks (GNNs) are the predominant models for representing molecular graphs. However, they are restricted to capturing pairwise interactions. Topological Neural Networks (TNNs) incorporate higher-order relational information beyond pair-
wise interactions, enabling richer representations than GNNs. GNNs also cannot compute important properties such as number and length of cycles (e.g., rings in the molecules), and have been augmented with persistence homology (PH) to mitigate this issue.
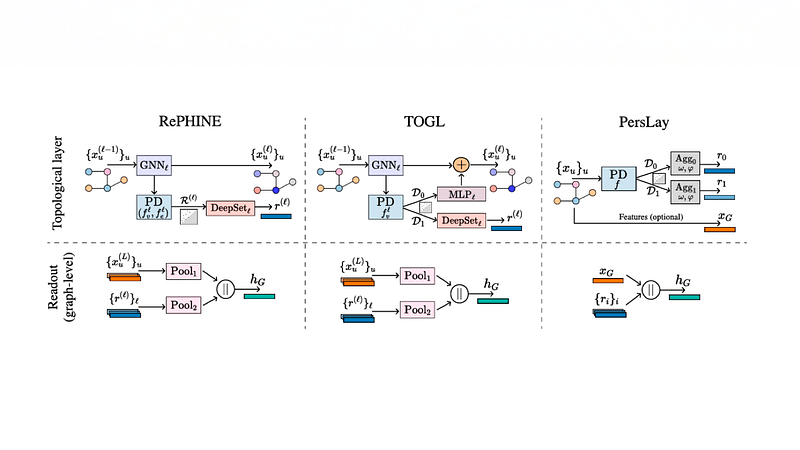
In an ICML 2024 paper, we design TopNets – a new class of deep learning models that is strictly more powerful than TNNs/GNNs and PH. TopNets can also be readily adapted to handle (symmetries in) geometric complexes, extending the scope of TNNs and PH to spatial settings (such as generating molecular conformations). TopNets achieve strong performance across diverse drug discovery tasks, including antibody design, molecular dynamics simulation, and drug property prediction.